Phenylketonuria - klassiske tegn på arvelig transmission og diætterapi
Indhold
- 1Hvordan manifesterer phenylketonuria
- 2Mekanisme for sygdomsudvikling
- 3Phenylketonuria hos børn
- 4Symptomer på sygdommen
- 5Årsager og triggere
- 6Diagnose
- 7Behandling af klassisk phenylketonuria
- 8Funktioner ved ernæring af nyfødte ogdiætterapi
- 9Diæt til børnehaver og skolebørn
- 10Produktgrupper med PKU
- 11Sådan kontrolleres niveauet af phenylalanin i blodet
- )12Video
Sygdomme, hvis forekomst er relateret til defekter i det genetiske cellulære apparat - phenylketonuria - er inkluderet i en lille liste over arvelige sygdomme, der kan behandles.Pioner for denne lidelse var en norsk læge IA Felling, det blev senere opdaget, at et enkelt gen kaldet phenylalaninhydroxylase-genet (den lange arm i det 12. kromosom indeholdende op til 4,5% af alt cellulært DNA-materiale) var ansvarlig for udviklingen og forløbet af sygdommen.Arvelig defekt fører til delvis eller fuldstændig deaktivering af leverenzymet fenylalanin-4-hydroxylase.
Hvordan fenylketonuri-sygdom opdages
Inherited phenylketonuria (PKU) sygdom fører til kronisk forgiftning af kroppen med giftige stoffer dannet som et resultat af nedsat aminosyremetabolisme og -proceshydroxylering af phenylalanin.Permanent rus forårsager skade på centralnervesystemet (CNS), hvis manifestation er en progressiv nedgang i intelligens (phenylpyruvic oligophrenia).
Fellings sygdom manifesteres i den overdrevne ophobning i kroppen af phenylalanin og produkterne fra dens forkerte stofskifte.Andre faktorer i udviklingen af phenylketonuria inkluderer nedsat aminosyretransport over blod-hjerne-barrieren, lave neurotransmittertællinger (serotonin, histamin, dopamin).I mangel af rettidig behandling af sygdommen fører til mental retardering og kan forårsage barnets død.

Mekanismen for sygdomsudvikling
Årsagen til genforstyrrelser er en metabolisk blok, der forhindrer dannelse af phenylalanin-4-hydroxylase (et enzym,som er ansvarlig for omdannelsen af phenylalanin-aminosyren til tyrosin).Proteinogen aminosyre-tyrosin er en integreret del af proteiner og melaninpigment, så det er et nødvendigt element for funktionen af alle kropssystemer, og dens mangel fører til fermentopati.
Inhiberingen af dannelse af metabolit forårsaget af mutationsinaktivering af enzymet er aktivering af hjælpemetaboliske forbindelser til phenylalanin.Aromatisk alfa-aminosyre, som et resultat af mangelfulde metaboliske processer, opdeles i toksiske derivater, som under normale betingelser ikke danner:
- phenylpyruvinsyre (phenylpyruvat) - en fedt-aromatisk alfa-keto syre, dens dannelsefører til myelination af neuronale processer og demens;
- phenylmelkesyre er et produkt dannet under genvindingen af phenylpyruvinsyre;
- phenylethylamin - den indledende forbindelse til biologisk aktive transmittere af elektrokemiske impulser, øger koncentrationen af dopamin, adrenalin og norepinephrin;
- Orthophenylacetat er et giftigt stof, der forårsager metabolske forstyrrelser i hjernen.
Medicinsk statistik viser, at der er et patologisk ændret gen i 2% af befolkningen, men det manifesterer sig ikke på nogen måde.Den genetiske defekt overføres til barnet fra forældrene kun i nærvær af sygdommen hos begge parter, hvor spædbarnet i 50% af tilfældene bliver bæreren af det muterede gen og forbliver sundt.Sandsynligheden for, at fenylketonuri hos nyfødte fører til sygdom, er 25%.
Efter hvilken type der er arvet
Fells sygdom er en genetisk abnormalitet, der er arvet fra en autosomal recessiv type.Denne type arv betyder, at udviklingen af tegn på en medfødt sygdom kun vil ske, når en mangelfuld genkopi fra begge forældre, der er heterozygote bærere af det ændrede gen, arves fra barnet.
Udviklingen af en medfødt sygdom i 99% af tilfældene skyldes en mutation af genet, der er ansvarligt for kodning af enzymet, der tilvejebringer syntese af fenylalanin-4-hydroxylase (klassisk phenylketonuria).Op til 1% af genetiske sygdomme er relateret til mutationsændringer, der forekommer i andre gener, der forårsagermangel på dihydropteridinreduktase (PKU type II) eller tetrahydrobiopterin (PKU type III).
Phenylketonuria hos børn
Den klassiske form for genetisk sygdom hos børn manifesterer sig i de fleste tilfælde udadtil, og starter ved 3-9 måneders alder.Nyfødte med et defekt gen, ser sunde ud, et særpræg er babyens specifikke vane (udseende).Den udtrykte symptomatologi vises i 6-12 måneder efter fødslen.
Type II PKU er kendetegnet ved, at de første kliniske symptomer optræder 1,5 år efter fødslen.Tegn på sygdommen forsvinder ikke efter diagnosen genetiske abnormiteter og begyndelsen af diætterapi.Denne type medfødt sygdom fører ofte til et dødeligt resultat i 2-3 år af et barns liv.De mest almindelige symptomer på type II PKU er:
- alvorlige psykiske lidelser;
- hyperrefleksi;
- nedsat motorisk funktion af alle ekstremiteter;
- syndrom med ukontrollerede muskelsammentrækninger.
Kliniske tegn på mutationsændringer i type III-gener ligner type II-sygdom.Tetrahydrobiopterinmangel er kendetegnet ved en triade af specifikke symptomer:
- en høj grad af mental retardering;
- størrelsen af kraniet reduceres tydeligt i forhold til andre dele af kroppen;
- muskelspasticitet (fuldstændigt tab af lemmemobilitet er muligt).

Manifestationer af Fellings sygdom
I kliniske forsøg og observationer er det blevet antydet, attoksiske derivater af phenylalaninmetabolisme forårsager et fald i intellektuel evne, som er progressiv og kan føre til demens (oligofreni, idioti).Blandt de sandsynlige årsager til irreversible forstyrrelser i hjerneaktiviteten af de mest rimelige anses for at være forårsaget af et fald i niveauet for tyrosinmangel på neurotransmittere, der transmitterer impulser mellem neuroner.
Det nøjagtige årsags- og virkningsforhold mellem arvelig sygdom og hjerneforstyrrelser er endnu ikke identificeret, ligesom mekanismen for udvikling skyldes phenylketonuri i sådanne mentale tilstande som ekkopraksi, ekkolalia, hårde angreb og irritabilitet.Resultaterne af analysen indikerer, at phenylalanin har en direkte toksisk virkning på hjernen, hvilket også kan forårsage et fald i intelligens.
Struktur og fænotype egenskaber
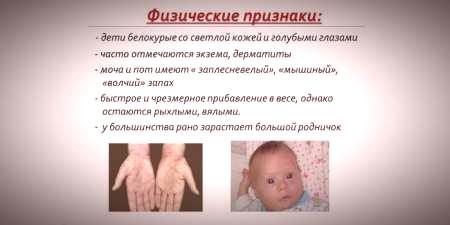
I betragtning af at hud- og hårpigmentmætning er afhængig af tyrosiniveauet i hepatocytmytokondrier, og phenylketonuri fører til stop af konventionen af fenylalanin, har patienter med denne sygdom særegenhederrecessive tegn).Forøget muskeltone medfører, at der forekommer afvigelser i kroppens struktur - det bliver dysplastisk.Karakteristiske ydre træk ved phenylketonuri inkluderer:
- hypopigmentering - lys hud, lyseblå øjne, misfarvet hår;
- cyanose af ekstremiteter;
- reduceret hovedstørrelse;
- kroppens specifikke position - når barnet prøver at stå eller sidde, vedtager barnet en "skræddersyet" stilling (arme og ben bøjede ved leddene).
Symptomersygdom
Med rettidig opdagelse behandles Fellings sygdom med succes ved at justere ernæring, og barnets udvikling sker i henhold til hans aldersgruppe.Problemer med at detektere en genmutation er, at tidlige tegn er vanskelige at påvise, selv af en erfaren børnelæge.Alvorligheden af symptomerne på medfødt sygdom øges, når barnet bliver ældre, fordi forbruget af proteinmad bidrager til udviklingen af CNS-lidelser.
Tegn hos nyfødte
I de første dage af barnets liv er tegn på unormale abnormiteter vanskelige at opdage - babyen opfører sig naturligt, der er ingen udviklingsforsinkelser.Symptomerne på sygdommen begynder at vises første gang 2-6 måneder efter fødslen.Forældrene skal være opmærksomme på babyens adfærd, der er kendetegnet ved lav aktivitet, sløvhed eller omvendt angst, hyper-excitability.
Med amningens begyndelse begynder nyfødte med mælk at komme ind i den nyfødte krop med mælk, som er en katalysator for udseendet af de første tegn, hvilket tydeligt indikerer, at sygdommen er begyndt at udvikle sig.Specifikke kliniske manifestationer af sygdommen inkluderer:
- konstant opkast (ofte betragtet som medfødt indsnævring af målmanden);
- hyppig opkast;
- intet svar på eksterne stimuli;
- muskeldystoni (reduceret muskelspænding);
- krampesyndrom (kramper af epileptisk eller ikke-epileptisk art).
Symptomer hos børn efter 6 måneder
Hvis manifestationen af genetisk sygdom ikke erforekom (eller ikke blev observeret) i de første 6 måneder efter fødslen af barnet, og efter denne periode er det allerede muligt nøjagtigt at bestemme forsinkelsen i psykomotorisk udvikling.Symptomer på genetiske lidelser forårsaget af enzymmangel hos børn over 6 måneder er:
- nedsat aktivitet (op til fuldstændig ligegyldighed);
- manglende selvbeherskelse, siddepladser;
- en særegen "mus" lugt i huden (lugten af skimmel skyldes udskillelsen af giftige phenylalaninderivater gennem svedkirtler og urin);
- tab af evne til visuelt at genkende forældres ansigter;
- afskalning af huden;
- dermatitis, eksem, scleroderma.
Fremskridt af sygdommen i fravær af behandling i barndommen
Hvis der ikke blev påvist udviklingsafvik i barndommen, og passende behandling ikke blev udført,sygdommen begynder at udvikle sig aktivt og fører ofte til handicap.Mangel på terapi i et tidligt stadie af sygdommen får følgende symptomer til at udvikle sig i en alder af 1,5 år:
- mikrocephaly (formindsket hjernestørrelse);
- prognathia (forskydning af den øvre tandpræstation fremad);
- sene tænder;
- emaljehypoplasi (udtynding eller fuldstændig fravær af tandemalje);
- forsinkelsen i sprogudviklingen indtil det fulde fravær af tale;
- 3, 4 oligofreni (mental retardering, mental retardering);
- medfødte hjertefejl (defekter i hjertemuskulaturen, dele af hjertet,store fartøjer);
- forstyrrelser i det autonome system (akrocyanose, overdreven sveden, arteriel hypotension);
- forstoppelse.
Årsager og provokerende faktorer
Til manifestationen af en autosomal recessiv mutation skal et defekt gen arves fra begge forældre.Genetiske sygdomme af denne type forekommer med samme hyppighed hos nyfødte drenge og piger.Patogenese af PKU er forårsaget af nedsat metabolisme af phenylalanin, som kan forekomme i 3 former.Diætterapi underkastes kun klassisk phenylketonuri type I.
Atypiske former for sygdommen kan helbredes ved at justere ernæring.Disse afvigelser er forårsaget af en mangel på tetrahydropterin, dehydropterinreduktase (sjældent - pyruvyltetrahydropterinsynthase, guanosin-5-triphosphat og andet).De fleste tilfælde af dødelige tilfælde registreres blandt patienter med sjældne variationer af PKU, med de kliniske manifestationer af alle former for sygdommen ens.Risikoen for at få en baby med et muteret phenylalaninhydroxylase-gen øges, hvis forældrene er nære slægtninge (i tæt beslægtede ægteskaber).
Diagnose
I tilfælde af mistanke om genetiske forstyrrelser stilles diagnosen på grundlag af et sæt data, der er opnået fra undersøgelsen af den medicinske historie - genealogiske data, resultaterne af kliniske og medicinske genetiske undersøgelser.Til rettidig påvisning af medfødte sygdomme (PKU, cystisk fibrose, galactosæmi osv.) Blev der udviklet et obligatorisk masseprogramlaboratorieundersøgelse af alle nyfødte (nyfødt screening).
Hvis forventningsfulde forældre er opmærksomme på bæreren af det muterede gen, tilbyder moderne medicin måder at påvise en defekt på graviditetsstadiet (prenatal diagnose af fosteret ved invasiv metode).Til opdeling af fenylketonuri i typer efter sværhedsgrad bruges en betinget klassificering baseret på niveauet af phenylalanin i en fibrinogenfri blodplasmavæske:
- Tung phenylketonuria - 1200 μmol /l.
- Gennemsnit - 60-1200 µmol /l.
- Lys (ingen behandling krævet) - 480 μmol /L.
Screeningstest
Påvisningen af genetiske abnormiteter finder sted i flere trin.På den første fase på barselhospitalet udtages alle spædbørn i 3-5 dages levetid perifert blod (fra fem) til undersøgelse.Materialet påføres på papirformular og sendes til det biokemiske laboratorium, hvor det er biokemisk analyse.I det andet trin i screeningstesten bestemmes det ved korrespondance mellem koncentrationen af den normale phenylalaninværdi.

Hvis der ikke registreres patologiske ændringer, udføres diagnosen, og barnets kort registreres.I tilfælde af afvigelser fra normen sendes resultaterne af diagnosen til en børnelæge for at sikre en mere nøjagtig undersøgelse af den nyfødte blodprøve.Barnets helbred afhænger af rettidig og nøjagtig implementering af alle foranstaltninger til at opdage afvigelser.Hvis diagnosen bekræftes efter gentagen screening, gør barnets forældre detsendt til en klinik til pædiatrisk genetik til behandling.
Analyser og undersøgelser til bekræftelse af diagnose
Gendiagnosticering efter detektion under den primære screeningstest af abnormiteter udføres ved at indsende prøverne igen.Ud over at bestemme indholdet af phenylalanin i blodet til metoder til diagnosticering af PKU hos børn og voksne inkluderer:
- Fældetest - bestemmelse af phenylpyruvinsyre i urinen ved at tilsætte jernchlorid til biomaterialet (farvning i blågrøn farve);
- Guthrie-test - vurdering af graden af mikroorganismeres respons på metabolismen eller enzymerne indeholdt i patientens blod;
- kromatografi - undersøgelsen af de kemiske egenskaber hos stoffer fordelt mellem to faser;
- fluorimetri - bestråling af biomateriale ved monokromatisk stråling for at bestemme koncentrationen af stoffer indeholdt deri;
- elektroencefalografi - diagnose af hjernens elektriske aktivitet;
- Magnetisk resonansafbildning er excitation af atomkernerne i celler ved hjælp af elektromagnetiske bølger og måling af deres respons.
Behandling af klassisk phenylketonuri
I hjertet af fenylketonuri-terapi er en begrænsning af forbruget af produkter, der er kilden til dyre- og vegetabilske proteiner.Den eneste metode til vellykket behandling er diætterapi, hvis tilstrækkelighed vurderes med indholdet af phenylalanin i serum.Maksimumsniveauet af aminosyrer hos patienter i forskellige aldersgrupperer:
- hos spædbørn og børn op til 3 år - op til 242 µmol /l;
- i børnehaver - op til 360 µmol /l;
- hos patienter i alderen 7 til 14 år - op til 480 μmol /l;
- hos unge - op til 600 μmol /l.
Diætets effektivitet afhænger af hvilket stadie af sygdommen kosten er korrigeret for.Ved tidlig diagnose af medfødt patologi ordineres diætterapi fra den 8. leveuge (efter denne periode begynder allerede irreversible ændringer).Fravær af rettidige foranstaltninger fører til komplikationer og et fald i intelligensniveauet med 4 point i 1 måned fra fødslen til behandlingsstart.

I betragtning af at den terapeutiske diæt for phenylketonuria giver fuldstændig udelukkelse fra kosten af animalsk protein, er der et behov for at bruge andre kilder til essentielle aminosyrer, såvel som vitaminer i gruppe B, calcium -og fosforholdige mineralforbindelser.Produkterne, der skal anvendes som proteinfrie kosttilskud, inkluderer:
- proteinhydrolysater (Amigen, Aminazol, Fibrinosol);
- indeholder ikke phenylalaninblandinger mættet med essentielle aminosyrer - Tetrafen, Phenylfri.
Foruden kurative foranstaltninger til at eliminere årsagen til nedsat funktion i kroppen, bør symptomatisk behandling udføres med det formål at eliminere talefejl og normalisere koordination af bevægelser.Kompleks terapi inkluderer fysioterapiprocedurer, massage, hjælp af en logoped, en psykolog, udførelse af gymnastiske øvelser.I nogle tilfælde i forbindelse med diætterapiviser brugen af anticonvulsiva, nootropiske og vaskulære medikamenter.
Særegenheder ved behandling af atypiske former
Phenylketonuria type II og III kan ikke behandles med lavproteindiet - niveauet af phenylalanin i blodet forbliver uændret, mens begrænsningen af proteinstrømmen ind i kroppen eller kliniske symptomer skrider frem, selv med faldet i aminosyreniveauet.Effektiv terapi af disse former for sygdommen udføres ved hjælp af:
- tetrahydrobiopterin - faktor af det berørte enzym;
- syntetiske analoger af tetrahydrobiopterin - disse stoffer trænger bedre ind i blod-hjerne-barrieren;
- substitutionsterapimedicin - fjerner ikke årsagen til phenylketonuri, men understøtter den normale funktion af kroppen (Levodopa sammen med Carbidofa, 5-oxytryptophan, 5-formyltetrahydrofolat);
- hepatoprotectors - understøtter leverfunktion;
- anticonvulsiva;
- introduktion af phenylalaninhydroxylase-genet i leveren er en eksperimentel metode.
Nyfødt ernæring og diætterapi
Amning er tilladt i det første leveår af en baby med PKU, men bør begrænses.Op til 6 måneder er det acceptable indtag af phenylalanin 60-90 mg pr. 1 kg babyvægt (100 g mælk indeholder 5,6 mg phenylalanin).Fra 3 måneder bør babyens diæt gradvist udvides, hvor der indføres frugtsaft og puréer i det.
Børn fra 6 måneders alder får tilsat kosten af vegetabilske puréer, grød (sago), proteinfriKiselev.Efter 7 måneder er det muligt at give baby lav-protein pasta, fra 8 måneder - brød, som ikke indeholder protein.Den alder, hvor proteinet i det syge barns krop skal begrænses, er ikke fastlagt.Læger diskuterer stadig gennemførligheden af livslang diætterapi, men de er enige om, at en diæt på mindst 18 bør følges.
Phenylketonuria, der er diagnosticeret hos en kvinde, er ikke en grund til at nægte et barns fødsel.Ekspanderende mødre med PKU for at forhindre fosterskade under graviditet og for at forhindre mulige komplikationer er det nødvendigt at observere en diæt med en phenylalanin-begrænset diæt (på leveringstidspunktet) (op til 242 μmol /l).
Laktosefri blandinger til babyer
Diætet til phenylketonuria er baseret på en betydelig reduktion i mængden af naturligt protein i den daglige diæt, men spædbarnets krop kan ikke udvikle sig normalt i mangel af de nødvendige sporstoffer.For at imødekomme babyens behov for protein bruges lactosefri aminosyreblandinger, som ifølge russisk lov skal leveres gratis.
Spædbarnets tolerance over for phenylalanin ændres hurtigt i det første leveår, så dets koncentration i babyens blod skal overvåges og kosten justeres.Blandingerne er designet til visse aldersgrupper:
- babyer under et år er ordineret Afenilak 15, Analog-SP, PKU-1, PKU-mix, PKU Anamix;
- Børn over 1 årår, udnævne beriget med vitaminer og mineraler blandinger med højt proteinindhold - PKU Prima, P-AM Universal, PKU-1, PKU-2, Maximeid XP, Maximum XP.

Proteintilskudsfødevarer
En af hovedkomponenterne i en diæt til phenylketonuria er produkter med lavt proteinindhold.Disse kosttilskud indeholder kaseinhydrolysat, tryptophan, tyrosin, methionin, nitrogen og giver babyens daglige behov for det protein, der er nødvendigt til normal udvikling og vækst.Specialiserede fødevarer, der fylder manglen på essentielle mineraler og aminosyrer, når de mangler i kosten, er:
- Berlofen;
- Zimorgan;
- Minafen;
- Aponti.
Diæt til børnehaver og skolebørn
Da kroppen tilpasser sig til phenylalanin, kan børn fra 5 år gradvist reducere deres kostbegrænsninger.Udvidelse af en diæt sker ved introduktion af korn, mejeriprodukter, kødprodukter.Ældre studerende har allerede en høj tolerance over for fenylalanin, så i denne alder kan du fortsætte med at udvide kosten, mens du overvåger responsen på eventuelle ændringer i ernæring.Følgende metoder bruges til at kontrollere barnets tilstand:
- vurdering af neurologiske parametre, psykologisk tilstand;
- kontrol af elektroencefalogrampræstation;
- bestemmelse af phenylalanin-niveau.
Grupper af fødevarer med PKU
Diætet til patienter med PKU sammen med lavt proteinstivelsesholdige produkter og medicinske blandinger inkluderer også produkter af naturlig oprindelse.Ved udarbejdelse af menuen skal mængden af forbrugt protein beregnes klart, og den af lægen anbefalede dosis bør ikke overskrides.For at fjerne toksiske virkninger på kroppen er der udviklet 3 lister over produkter, der indeholder forbudte (røde), ikke anbefalede (orange) og tilladte (grønne) genstande.
Rødliste
Phenylketonuria udvikler sig på baggrund af fraværet af et enzym, der omdannes til tyrosinphenylalanin, så et højt proteinindhold er grundlaget for listen over produkterne på den forbudte (røde) liste.Elementerne på denne liste skal fuldstændigt udelukke kosten for en patient med PKU:
- kød;
- indre organer i dyr, biprodukter;
- pølser, pølser;
- skaldyr (inklusive fisk);
- æg fra alle fugle;
- mejeriprodukter;
- nødder;
- bælgplanter og korn;
- sojaprodukter;
- gelatineholdige skåle;
- konfekture;
- aspartam.
Orange liste
Produkter, der skal doseres til et barn, der er diagnosticeret med PKU, er inkluderet på den orange liste.Inkluderingen i kosten af varerne på denne liste er acceptabel, men i et strengt begrænset antal.Selvom disse produkter ikke indeholder meget protein, kan de også øge niveauet af phenylalanin, så deres anvendelse anbefales ikke:
- konserverede grøntsager;
- kartoffel- og risfad;
- kål;
- mælk;
- sherbet.
Grøn liste
Proteinfrie produkter må bruges til patienter med en fenylketonuri-diagnose uden begrænsning.Før du køber varerne på den grønne liste, er det nødvendigt at undersøge sammensætningen, der er angivet på pakningen, og sørge for, at der ikke er noget farvestof til aspartam, der indeholder phenylalanin:
- frugter;
- grøntsager (undtagen kartofler og kål);
- bær;
- greener;
- stivelsesholdig korn (sago);
- honning, sukker, marmelade;
- melprodukter fremstillet af majs eller rismel;
- olier, fedtstoffer (smør, solsikke, oliven).
Hvordan man kontrollerer niveauet af phenylalanin i blodet
Phenylketonuria er en uhelbredelig sygdom, der kan overføres til stagnationsfasen ved brug af diæt og terapeutiske foranstaltninger.Ved ændring af levevilkår, forstyrrelse af en diæt, kan sygdommen forværres igen, derfor har patienter brug for livslang observation.Kontrolprocessen er periodisk at bestemme niveauet af phenylalanin i blodet.Analysefrekvensen afhænger af patientens alder:
- op til 3 måneder - blodscreening bør udføres ugentligt for at opnå ensartede resultater;
- fra 3 måneder til 1 år - 1-2 gange om måneden;
- 1 til 3 år - en gang hver anden måned;
- ældre end 3 år - kvartalsvis.

Blod til analyse gives 3-4 timer efter indtagelse.Ud over screening styres udviklingen af PKU ved at bestemme ernæringsstatus, fysisk, følelsesmæssig udvikling af patienten, niveauet for intellektuelle evner.og sprogudvikling.Observationer kan kræve yderligere diagnosticering med involvering af passende specialister.
Video
Oplysningerne i denne artikel er kun vejledende.Artiklen kræver ikke selvbehandling.Kun en kvalificeret læge kan diagnosticere og anbefale behandling baseret på de individuelle egenskaber hos den pågældende patient.















